Current Lab Research
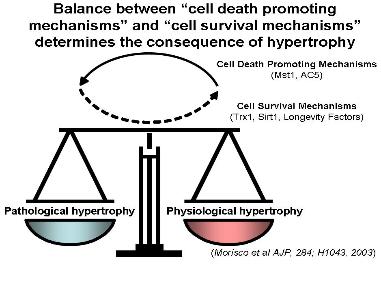


Cardiac hypertrophy is the enlargement of cardiac myocytes, and is observed in many forms of heart disease. Nevertheless, increasing lines of evidence suggest that the progression of heart failure is determined by the balance between cell death promoting mechanisms and cell survival/protective mechanisms, rather than by the presence of cardiac hypertrophy alone (Figure 1). This laboratory studies the signaling mechanisms which regulate the growth and death of cardiac myocytes, with a particular emphasis on those relevant to the pathogenesis of heart Transgenic mice and knock-out (KO) mice are routinely generated, and the laboratory has independent setups for ES cell culture and mouse surgery/physiology. Genomic and proteomic analyses are routinely conducted through in-house collaborations.
One of the major subjects of study in this laboratory is the function of mammalian sterile 20 like kinase (Mst1), a serine threonine kinase, in the heart. We have identified Mst1 as one of the most prominent kinases activated when cardiac myocytes undergo apoptosis. Using transgenic approaches, we have shown that Mst1 strongly activates apoptosis in cardiac myocytes and that endogenous Mst1 is involved in ischemia/reperfusion injury (1) and the development of heart failure after myocardial infarction (2) in the mouse heart. Unexpectedly, we found that Mst1 is also involved in many other functions in the heart, such as inhibition of compensatory hypertrophy, induction of endoplasmic stress and mitochondrial dysfunction, all of which are intimately involved in the pathogenesis of heart failure. Recent evidence suggests that Mst1 belongs to an evolutionarily conserved signaling cascade regulating organ size, which has been most extensively studied in Drosophila and is called the “hippo” pathway. We have shown that Mst1 activates Lats2, a homologue of Drosophila Wts, which in turn plays a critical role in regulating the size of the heart (3). We expect that both upstream regulators and downstream effectors of Mst1 and their functions in mammalian cells, including cardiac myocytes, will be identified in the next few years (Figure 2).
We also investigate the function of thioredoxin 1 (Trx1) in the heart. Trx1 is a 12kD anti-oxidant which reduces proteins with disulfide bonds through a thiol-disulfide exchange reaction (Figure 3). Trx1 is activated by stress and plays a protective role in the heart (4). We have shown that Trx1 negatively regulates pathological hypertrophy (5) and increases mitochondrial function through increased expression of genes involved in the TCA cycle and (7). In collaboration with the proteomic core facility, we are actively investigating molecular targets of Trx1.
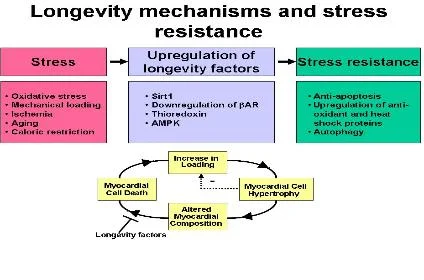
Another important subject of study is the role of longevity factors in mediating cardioprotection in the heart (8). In lower organisms, activation of molecular mechanisms mediating extension of lifespan confers stress resistance to the organism. We hypothesize that activation of known longevity mechanisms in the heart may make the heart more stress resistant (Figure 4) (9). Sirt1 is an NAD+-dependent class III histone deacetylase which plays an important role in mediating lifespan extension in response to caloric restriction in lower organisms. In the heart,
Sirt1 is upregulated by stress, and mild to moderate expression of Sirt1 retards aging of the heart and increases the heart’s resistance to oxidative stress (10). We are currently focusing on the molecular functions of Sirt1 and Sirt3 in the heart, as well as studying the roles of other longevity mechanisms, such as Trx1, adenylyl cyclase type 5 KO and AMP-dependent protein kinase (AMPK).


Whereby cytoplasmic proteins and organelles are degraded and recycled through lysosomes (11). Interestingly, autophagy is required for lifespan extension in response to dietary stress in C elegans, suggesting that autophagy could be yet another example in which a lifespan extension mechanism induces protection against stress. In the heart, basal levels of autophagy play a homeostatic role, and the absence of autophagy causes cardiac dysfunction and the development of cardiomyopathy. On the other hand, myocardial ischemia and reperfusion elicit conditions which strongly induce autophagy, including energy starvation, damage to intracellular organelles, protein aggregation, oxidative tress and ER stress (Figure 5). Using a mouse model of ischemia/reperfusion, we are studying both the signaling mechanisms and the functional significance of autophagy in the heart. We have shown recently that, although induction of autophagy during the ischemic phase is
protective, further enhancement of autophagy during the reperfusion phase may induce cell death and appears to be detrimental in the heart (12, 13).
Finally, cardiac hypertrophy is regulated by negative, as well as positive, regulators (14). Studying the mechanisms by which the endogenous negative regulators inhibit hypertrophy would provide useful information regarding the pathogenesis of cardiac hypertrophy and heart treatments for heart failure. Glycogen synthase kinase-3 (GSK-3) is an important negative regulator of cardiac hypertrophy (15)
(Figure 6). We have on the contrasting cardiac functions of the GSK-3 isoforms, namely GSK-3a and GSK-3b, in the heart (17-19). In addition, we are also studying the effect of GSK-3 modulation upon mesenchymal stem cell differentiation into the cardiac myocyte lineage following myocardial infarction in vivo.